La beta-talassemia (β-talassemia) o anemia mediterranea è un gruppo di malattie ereditarie del sangue. Esse sono causate da una ridotta o assente sintesi delle catene beta dell'emoglobina; ciò può comportare risultati variabili, che vanno da una grave anemia a una condizione clinicamente asintomatica. L'incidenza annua globale è stimata in 1 su 100 000. La beta-talassemia è una forma di talassemia causata dalle mutazioni nel gene HBB sul cromosoma 11, ereditato in modo autosomico recessivo. La gravità della malattia dipende dalla natura della mutazione.
L'anemia mediterranea è di solito asintomatica nella forma eterozigote, mentre si manifesta nella forma omozigote recessiva; i sintomi compaiono a circa sei mesi di vita con pallore, febbre, debolezza e ritardo nello sviluppo osseo. L'anemia mediterranea provoca globuli rossi molto piccoli detti microciti con ridotta concentrazione di emoglobina. Buona parte dei precursori emopoietici muoiono per apoptosi a causa dell'eccesso di catene alfa (emopoiesi inefficace) o arriva a completare la maturazione per poi subire lisi nel torrente circolatorio (similmente alle anemie emolitiche).
Il blocco del gene HBB comporta una diminuzione della sintesi della catena beta dell'emoglobina. L'incapacità del corpo di costruire nuove catene porta alla sottoproduzione di HbA (formata da due catene alfa e due catene beta) in favore della produzione di HbA2, una forma di Hb formata da due catene alfa e due catene delta, invece di beta.
La riduzione di HBA totale disponibile a riempire i globuli rossi, a sua volta, porta all'anemia microcitica. L'anemia microcitica sviluppa infine un funzionamento insufficiente dei globuli rossi.
A causa di ciò, il paziente può necessitare di trasfusioni di sangue. Le ripetute trasfusioni portano ad accumulo di ferro, che si traduce nell'intossicazione da ferro, portando a sviluppare siderosi miocardica e scompenso cardiaco, che possono portare al decesso del paziente.
Epidemiologia
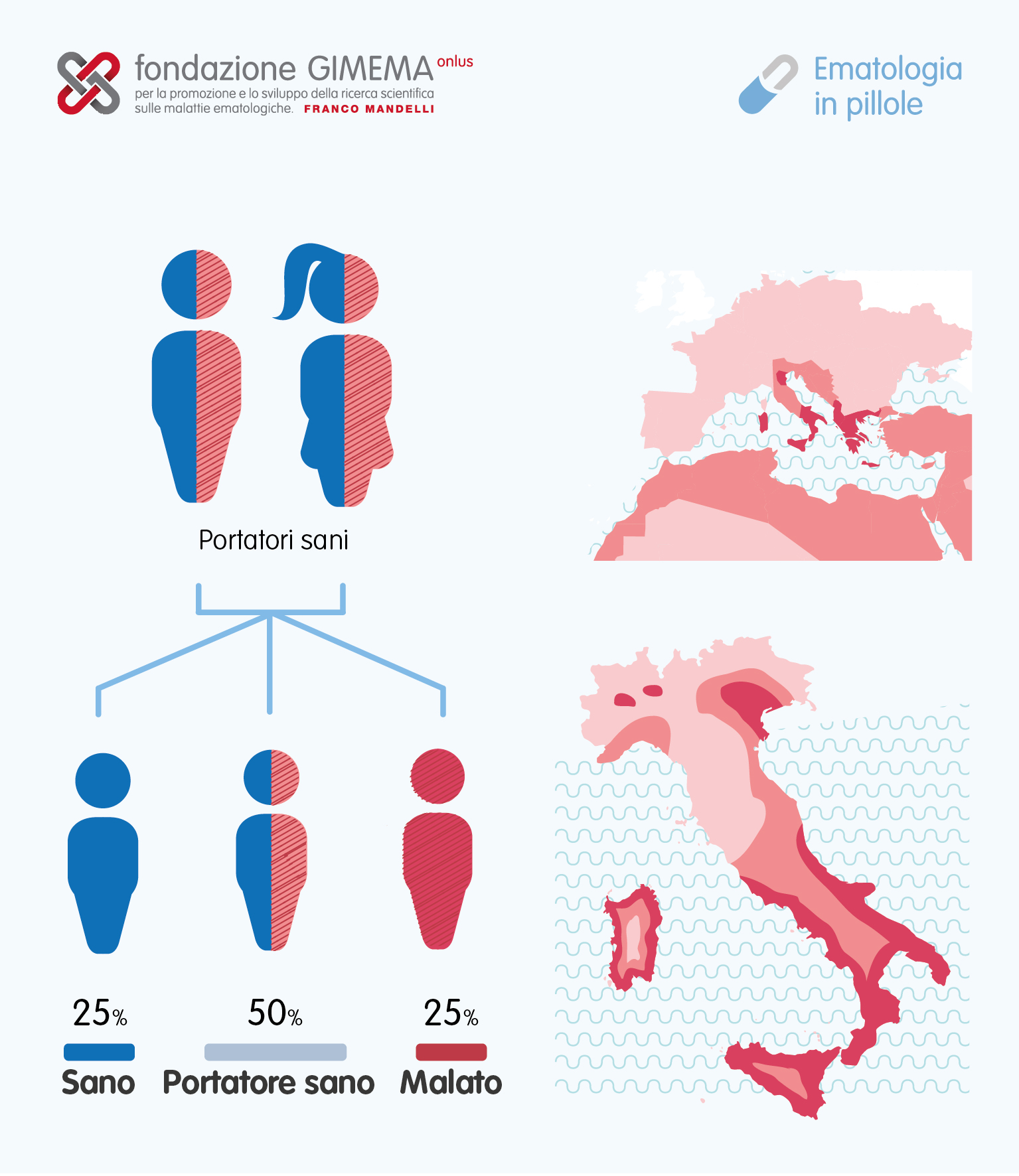
La versione beta di talassemia è particolarmente diffusa tra i popoli del Mediterraneo e questa associazione geografica è l'etimologia del suo nome: "thalassa" (θάλασσα) è la parola greca che indica il mare e "haema" (αἷμα) indica il sangue.
In Europa le più alte concentrazioni della malattia si trovano in Grecia e nelle regioni costiere turche. Le principali isole mediterranee (tranne le Baleari), come la Sicilia, la Sardegna, la Corsica, Cipro, Malta e Creta sono particolarmente colpite.
Altri popoli del Mediterraneo, così come quelli nelle immediate prossimità, presentano anch'essi alti tassi di incidenza, compresi gli individui provenienti dall'Asia occidentale e dal Nord Africa. I dati indicano che il 15% dei ciprioti greci e turchi è portatore di geni di beta-talassemia, mentre il 10% è portatore dei geni dell'alfa-talassemia.
Adattamento evolutivo
La talassemia può conferire un grado di protezione contro la malaria, che è, o è stata, diffusa nelle regioni in cui la talassemia è comune, conferendo un vantaggio di sopravvivenza selettiva (noto come vantaggio eterozigote), perpetuando così la mutazione. A questo proposito, le varie talassemie assomigliano a un'altra malattia genetica che colpisce l'emoglobina: l'anemia falciforme.
Eziologia
Mutazioni
Si possono distinguere due grandi gruppi di mutazioni:
- Forme non-deletion: questi difetti, in generale, comportano una sola sostituzione di base o una delezione o inserto vicino o a monte del gene beta-globina. Il più delle volte, le mutazioni avvengono nelle regioni promotrici precedenti i geni della beta-globina. Meno spesso, varianti anomale di splicing sono ritenute per contribuire alla malattia.
- Forme con delezione: delezioni di dimensioni diverse che coinvolgono il gene beta globina producono differenti sindromi quali βo o la sindrome della persistenza dell'emoglobina fetale.
Assemblaggio dell'mRNA
La beta-talassemia è una malattia ereditaria che colpisce l'emoglobina. Come con circa la metà di tutte le malattie ereditarie, vengono ereditati i danni dovuti alla mutazione del montaggio dell'RNA messaggero (mRNA) che viene trascritto a partire da un gene. Il DNA contiene sia le istruzioni (geni) per la corretta successione degli aminoacidi che andranno a formare le proteine sia tratti non codificanti che giocano ruoli importanti nel regolare i livelli di produzione delle stesse.
Nella talassemia, un'ulteriore regione non codificante di lunghezza contigua o un frammento discontinuo è inclusa nell'mRNA. Questo accade perché la mutazione annulla il confine tra le porzioni introniche ed esoniche. Per via della possibile presenza di tutte le sezioni di codifica, la normale emoglobina può essere sintetizzata, mentre il materiale aggiuntivo può essere causa di disfunzioni abbastanza gravi da scaturire in anemia. Ogni emoglobina possiede una porzione centrale di ferro (gruppo eme) che permette alla catena proteica di una subunità di piegarsi intorno. L'emoglobina normale di una persona adulta contiene due subunità alfa e due beta. Le talassemie tipicamente riguardano solo l'mRNA per la produzione delle catene beta (da qui il nome). Dal momento che la mutazione può essere un cambiamento in una sola base (un "polimorfismo a singolo nucleotide"), vi è un continuo sforzo nelle terapie geniche per riuscire fare quella singola correzione.
Clinica
Classificazione
Allo stesso genotipo può corrispondere una diversa gravità di malattia.
Segni e sintomi
Sono state descritte tre forme principali: la talassemia major, la talassemia intermedia e la talassemia minor. Tutte le persone con talassemia sono suscettibili di complicazioni di salute che coinvolgono la milza (che spesso risulta ingrandita e frequentemente viene rimossa) e calcoli biliari. Queste complicazioni si trovano principalmente nei pazienti con talassemia major e intermedia. Gli individui con beta-talassemia major di solito manifestano, entro i primi due anni di vita, grave anemia, scarsa crescita e anomalie scheletriche durante l'infanzia. Se non trattata, la condizione major alla fine porta alla morte, di solito per insufficienza cardiaca. Pertanto, lo screening alla nascita è molto importante.
L'eccesso di ferro provoca gravi complicazioni al fegato, al cuore e alle ghiandole endocrine. Gravi sintomi includono cirrosi epatica e, in casi estremi, il tumore al fegato. L'insufficienza cardiaca, l'insufficienza della crescita, il diabete e l'osteoporosi sono fattori che incidono sulla qualità e durata della vita nei pazienti con talassemia major. Le principali anomalie cardiache includono disfunzioni al ventricolo sinistro, ipertensione polmonare, aritmia, valvulopatia e pericardite. Un maggior assorbimento del ferro a livello gastrointestinale si nota in tutti i gradi di beta-talassemia e una maggiore distruzione di globuli rossi da parte della milza, a causa dell'eritropoiesi inefficace, determina un ulteriore rilascio di ferro nel sangue.
Diagnosi differenziale
Un dolore addominale causato da ipersplenismo e infarto splenico e dolore al quadrante destro superiore causato da calcoli biliari sono le principali manifestazioni cliniche. Tuttavia, i soli sintomi sono insufficienti per formulare una diagnosi di talassemia. I seguenti segni correlati possono attestare la gravità del fenotipo: pallore, scarsa crescita, insufficiente assunzione di cibo, splenomegalia, ittero, iperplasia mascellare, malocclusione dentale, colelitiasi e fratture patologiche. Sulla base dei sintomi, esami possono essere prescritti per arrivare alla diagnosi differenziale. Questi test includono un emocromo completo; l'elettroforesi dell'emoglobina; la transferrina, la ferritina, la capacità di rilegatura del ferro, l'urobilina, la bilirubina, lo striscio di sangue periferico.
Analisi del DNA
Tutte le beta-talassemie possono presentare globuli rossi anomali; attraverso l'analisi del DNA è possibile ricostruire una storia familiare. Questo test è utilizzato per indagare delezioni e mutazioni nei geni produttori di alfa e beta globina. Studi di famiglia possono essere condotti per valutare lo stato di portatore e il tipo di mutazioni presenti in altri membri della famiglia. Il test del DNA non è di routine, ma può aiutare a diagnosticare la talassemia e determinare lo stato di portatore. Nella maggior parte dei casi il medico curante utilizza una prediagnosi clinica per valutare i sintomi dell'anemia: affaticamento, mancanza di respiro e scarsa tolleranza allo sforzo. Ulteriori analisi genetiche possono includere la cromatografia liquida ad alta prestazione quando l'elettroforesi di routine potrebbe rivelarsi difficile.
Trattamento
Talassemia major
I bambini affetti richiedono regolari trasfusioni di sangue per tutta la vita e possono sviluppare complicazioni, soprattutto alla milza.
I pazienti che ricevono frequenti trasfusioni di sangue vanno incontro a un sovraccarico di ferro; il trattamento di chelazione del ferro è necessario per evitare danni agli organi interni: chelanti popolari includono la deferoxamina e il deferiprone. Lo svantaggio della deferoxamina è che talvolta il trattamento può essere doloroso e scomodo; inoltre, questo farmaco sembra essere associato a una precoce comparsa di presbiopia, pertanto sarebbe preferibile sostituirla con deferiprone o deferasirox. Il chelante orale deferasirox è stato approvato per l'uso nel 2005 in alcuni Paesi e offre migliori prestazioni a un costo superiore. I progressi nei trattamenti di chelazione permettono ai pazienti con talassemia major di vivere una vita lunga se possono avere accesso a un trattamento adeguato.
A questi soggetti si consiglia poi di limitare il consumo di carne, alimento notoriamente ricco di ferro in forma eme, e di bere tè, perché i tannini ivi contenuti fanno diminuire l'assorbimento del ferro nell'intestino.
Il ricorso al trapianto di midollo osseo può essere risolutivo per alcuni bambini, sempre tenendo conto della compatibilità genetica. Il trapianto è indicato per i pazienti con una grave talassemia major, e può eliminare la dipendenza dalle trasfusioni.
Talassemia intermedia
I pazienti possono necessitare di trasfusioni di sangue episodiche che comunque possono comportare un sovraccarico di ferro e dunque il paziente può necessitare della terapia chelante per rimuoverlo.
Talassemia minor
I pazienti vengono spesso monitorati senza un trattamento. Anche se non è pericolosa per la vita, la condizione può influire sulla qualità della vita causando anemia.
Prevenzione
Trattandosi di una eterozigosi, la prevenzione si applica per evitare l'insorgenza della beta-talassemia grave. È indispensabile che una persona con anemia mediterranea non abbia figli con un'altra persona con la stessa patologia per scongiurare il rischio di trasmissione di questa malattia ereditaria grave. Per evitare di avere figli con beta-talassemia sia in forma omozigote (molto grave) sia eterozigote (non grave, ma può causare astenia) viene sempre più spesso usata la diagnostica prenatale.
Nelle zone a maggior diffusione della malattia è prevista l'analisi di tutta la popolazione, in modo da individuare le persone affette da anemia mediterranea e informare loro sui rischi di trasmissione alla prole. Nelle altre zone, è affidato all'individuo affetto il compito di effettuare l'analisi sulla propria discendenza; generalmente, è indicato eseguire l'esame intorno all'età di 11 anni.
Note
Altri progetti
- Wikimedia Commons contiene immagini o altri file su anemia mediterranea
Collegamenti esterni
- Centro della Microcitemia di Roma, su blod.info.